 氮气还原耦合氢气氧化实现连续流电化学合成氨
氮气还原耦合氢气氧化实现连续流电化学合成氨
氨是合成化肥、药物和精细化学品的重要原料。目前,工业合成氨采用Haber-Bosch工艺,即氮气和氢气通过铁基催化剂,在高温高压(350-450℃,150-200 bar)的反应条件下合成氨。Haber-Bosch 工艺被认为是20世纪最具影响力的威廉希尔官方网站
成就之一。
2021年,通过Haber-Bosch工艺,全球氨产量高达1.82亿公吨。其中,约70%的氨用于了化肥生产,全球约50%的粮食生产依赖于氨衍生的化肥(Chem Catalysis, 2 (10), 2590-2613 (2022).)。以上足可见合成氨威廉希尔官方网站
对人类的影响之大,然而目前工业合成氨路线,生产1公吨氨释放约2.1公吨二氧化碳温室气体,而且需要大型工厂和高额的资本投资,该工艺的能耗就占全球能源消耗的1%,二氧化碳的排放量占全球排放总量的1.3%。
传统的合成氨威廉希尔官方网站
是密集型生产氨,化肥通过运输到达用户。目前,随着可再生电力价格持续降低,以可再生能源为驱动力,直接从氮气和水进行电化学合成氨是有潜力的合成氨路线之一。该过程可在较小规模的装置中,利用分散的可再生能源进行分散式地合成氨,预期会带来巨大的经济和社会效益,例如降低缺乏交通网络或基础设施的发展中国家和偏远地区的化肥价格。
近年来,电化学合成氨和电催化合成氨得到了科学界广泛的关注和探究,期望电化学合成氨路线能够取代或补充Haber-Bosch工艺。遗憾的是,2020年,Douglas R. MacFarlane课题组详细调研了127篇水系电催化合成氨的文章(Nat Commun 11, 5546 (2020).),氨的产量太低以致于不能确定所产的氨是否来自氮气还原,目前来看,水系电催化还原氮气合成氨路径的可行性仍然悬而未决。
2019年,Ib Chorkendorff教授课题组通过定量同位素和严格的气体纯化实验表明(Nature 570 (7762), 504-508 (2019).),在室温下电化学合成氨的可靠途径之一是锂介导氮气还原反应(Li-NRR)。 锂介导合成氨最早可追溯到1930年,Fichter等人使用LiBr和LiCl的醇溶液进行电化学合成氨(Helv. Chim. Acta 13, 1228–1236 (1930))。
1993年,Tsuneto等人进行了优化(Chem. Lett., 851-854 (1993)),使用四氢呋喃(THF)和乙醇(99/1体积比)作为溶剂,LiClO4作为电解质。1994,Tsuneto等人通过增加反应器压力,再次提升了氨的法拉第效率。自2019年以来,Li-NRR经历了快速发展的时期。
2022年,在20 bar条件下,在单室间歇性反应釜中,总电流密度已经实现1A/cm2(Joule 6, 2083-2101 (2022))。2022年,在15 bar条件下,在单室间歇性反应釜中,氨的法拉第效率已经接近100%(Nature 609, 722-727 (2022))。
尽管取得了较大进展,所合成的氨中的氮原子来自于氮气还原,但是所合成的氨中的氢原子来自于哪里?在已报道的Li-NRR研究中,几乎所有的报道都在阳极氧化有机溶剂作为质子源,合成氨不能以消耗有机溶剂为代价!基于此,Karthish Manthiram课题组(Nat. Catal. 3, 463-469 (2020))最早提出在阳极侧引入氢气氧化反应(HOR)来提供可持续的质子源,但是并未证明氨中的氢来自于HOR,而且电解池电压高达20-30V(未给出阳极电位),导致伪能量效率(EE)只有2.8%,该体系只能够稳定5-8 min。
Douglas R. MacFarlane课题组也曾试图正在阳极引入HOR(Science 372, 1187-1191 (2021)),在高压反应釜中通入0.5 bar H2和19.5 bar N2使用离子液体作为质子穿梭剂(proton shuttle),然而在单室间歇性反应釜,不仅存在H2传质限制,而且H2可以与金属锂反应形成氢化锂,这阻碍了锂在室温下活化氮气的能力。
目前为止,大部分的Li-NRR研究在单室间歇反应釜中进行,存在以下挑战:
(1)气体反应物传质限制:N2和H2必须溶于电解液才能参与反应。因此,通常增加压力来实现较高的产氨效率。
(2)间歇性产氨,难以放大生产。
(3)难以利用HOR作为可持续的质子源,H2会消耗金属锂。
在常温常压(1 bar和25 oC)下进行Li-NRR的挑战如下:
(1)低产氨法拉第效率和能量效率,在单室间歇反应釜中,FE和EE都低于10%。
(2)牺牲溶剂作为质子源。
(3)在装配气体扩散电极的电解池中,稳定性极差。
基于这些挑战和难题,丹麦科技大学Jens K. Nørskov教授和Ib Chorkendorff教授等人设计了有效面积为25 cm2的连续流电解池,采用规整孔尺寸的不锈钢网(SSC)作为气体扩散电极来避免传质限制,氮气还原(NRR)耦合氢气氧化(HOR),在常温常压条件下,实现了61%的法拉第效率,13%的能量效率。
开发了在有机溶剂中稳定催化HOR的PtAu合金催化剂,PtAu合金催化剂极大降低了阳极电位,避免了溶剂的氧化。通过原位同位素质谱实验(D2氧化),证明了氨中的氢来自于HOR,EtOH作为质子穿梭剂。
图文介绍
首先构建了一个三室连续流反应器,GDE位于气体室与电解液室之间,气体反应物直接供给在GDE电极的一侧,气体流道的面积是25 cm2。Li-NRR在连续流电解池中的过程如下:锂离子(Li+)通过固体电解质界面(SEI)从本体电解液扩散到阴极电极表面电化学还原为金属锂,随后金属锂与N2反应形成氮化锂 (LixNyHz),氮化锂通过质子穿梭剂(EtOH)质子化来连续产生氨。PtAu阳极催化剂上稳定地发生HOR为该过程提供可持续的质子源。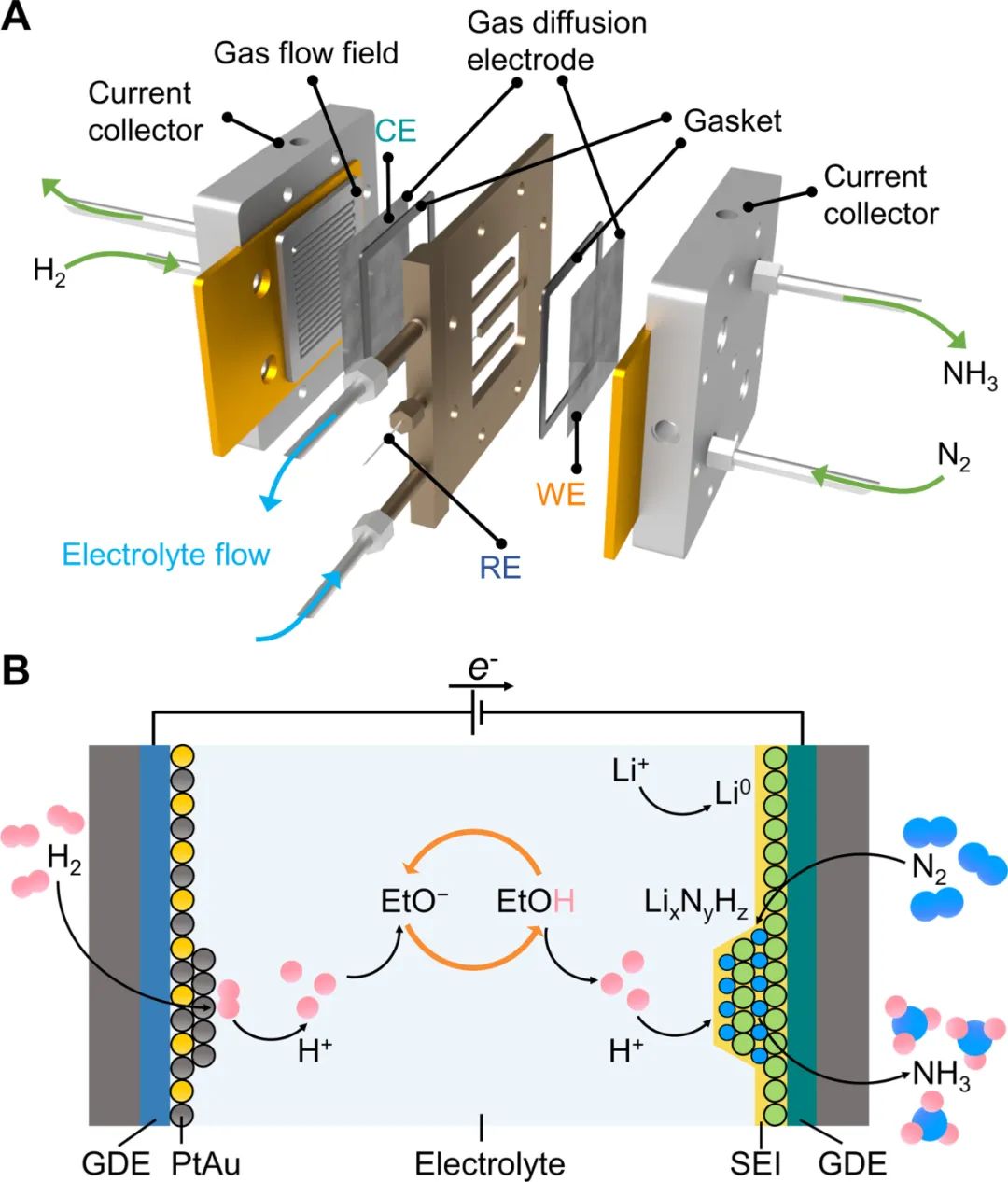
图1. 用于电化学合成氨的连续流反应器的示意图和Li-NRR过程示意图
目前为止,极少数文献研究了有机电解液中的HOR,与水系中的经典HOR不同,有机体系中的HOR催化剂非常容易失活,有机溶剂或反应中间体会毒化活性位点。在有机体系HOR中,Pt的催化活性在几分钟内失活。Douglas R. MacFarlane课题组探究了一系列在有机体系中的HOR催化剂,发现失活速率最小的PtRu催化剂在20 min左右也会失活(ACS Catal. 12, 5231-5246 (2022))。
因此,非常需要开发一种在有机体系中长期稳定的HOR催化剂。在小分子电氧化(甲酸/甲醇/乙醇)领域,Pt很容易被一氧化碳(CO)或其他中间体中毒。在Pt基双金属催化剂中,PtAu表现出良好的催化活性和抗CO中毒的特性。还有研究表明,用Au团簇修饰的Pt可极大增加氧还原反应的稳定性。受这些研究的启发,作者认为Au对抑制有机分子或中间体的吸附有积极作用,PtAu催化剂有可能成为有机体系中最稳定的HOR催化剂之一。
理论计算表明,与纯Pt表面相比,H、CO、THF和呋喃等物种在PtAu表面的吸附被削弱。H在PtAu表面上的吸附能比略弱于H在Pt表面的吸附能,从而使得PtAu的HOR反应速率比纯Pt高1-2个数量级(图2A)。与此同时PtAu表面上HOR相对于THF氧化反应的选择性比纯Pt表面高5-6个数量级,这显着地降低了THF氧化副反应的速率。
更重要的是,CO作为毒化Pt基金属的主要中间体,在Li-NRR反应中,其主要来源于乙醇氧化过程中碳碳键(C-C)的断裂。理论计算表明,C-C键在纯Pt表面断裂所需要的反应能垒在室温条件下极易克服,但是在PtAu表面,C-C键断裂需要克服较高的反应势垒所以不易发生, 从而抑制了CO的产生和表面活性位点的毒化。
通过氢气泡模板法在SSC基底上制备了Pt和PtAu电极(Pt/SSC和PtAu/SSC)。HOR性能是使用1M LiBF4的THF电解液在氩气手套箱中进行评估。在恒电流测试(CP)期间,Pt/SSC在几分钟内失活,阳极电位增加到1V vs. Pt,此电位下可发生THF氧化的副反应(图2B)。
相比之下,PtAu/SSC在5小时内电位保持在0.3V vs. Pt,证明了PtAu在有机体系HOR中的高活性和长期稳定性(图2B)。在对PtAu进行长期稳定性测试后,角分辨XPS结果表明Pt在PtAu催化剂表面富集(图2C)。DFT计算表明,即使是微量的呋喃、CO和甲基也会导致Pt原子偏析到表面(图2D),而且底层的Au进一步削弱了Pt覆盖层对有机物的吸附强度。
底层的Au促进了表面类Pt(111)覆盖层的形成,记作Pt(111)/Au。乙醇氧化中,C-C键断裂所需要的反应势垒在Pt(111)/Au表面的是在PtAu合金的1.5倍,而Pt(111)/Au上的HOR本征活性与PtAu相当。所以,PtAu作为真实催化剂Pt的前体,真实催化剂Pt(111)/Au表面在保有与PtAu合金相同的HOR本征活性同时,进一步抑制了CO的产生和表面活性位点的毒化。
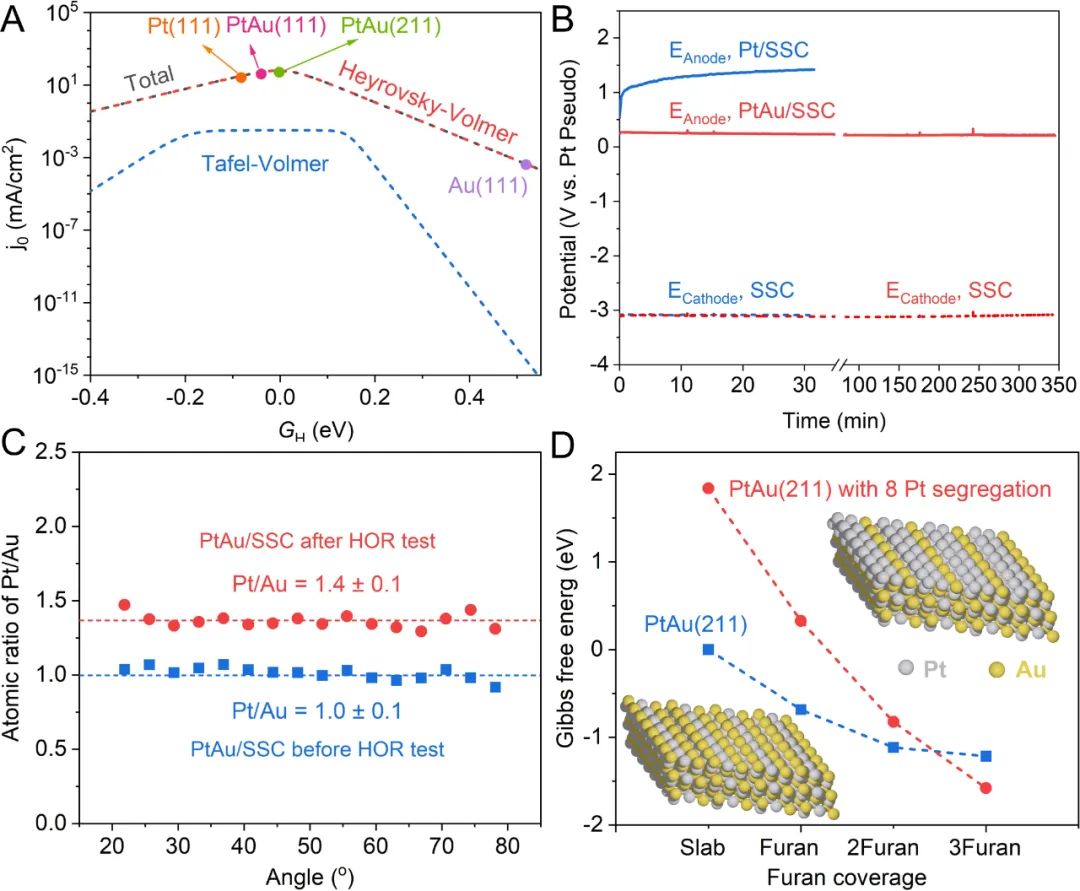
图2. HOR催化剂的理论和实验探究
为了证明NRR与HOR的耦合,在连续流电解池中进行了同位素原位质谱实验。当D2流过阳极气体室,测量阴极气体出口处的产物,原位质谱可将电解液反应产生的含H的产物和D2氧化产生的含D的产物区分开来,最初,阴极完全被新鲜的电解液覆盖,产生主要是含H的产物,如NH3和NH2D。
随着实验的进行,越来越多的含D产物形成(图3C),最终完全氘代的氨(ND3)被检测到。这有力地证明了NRR和HOR的耦合作用,所合成的氨中的氢来自于HOR,并证明了乙醇作为质子穿梭剂的能力。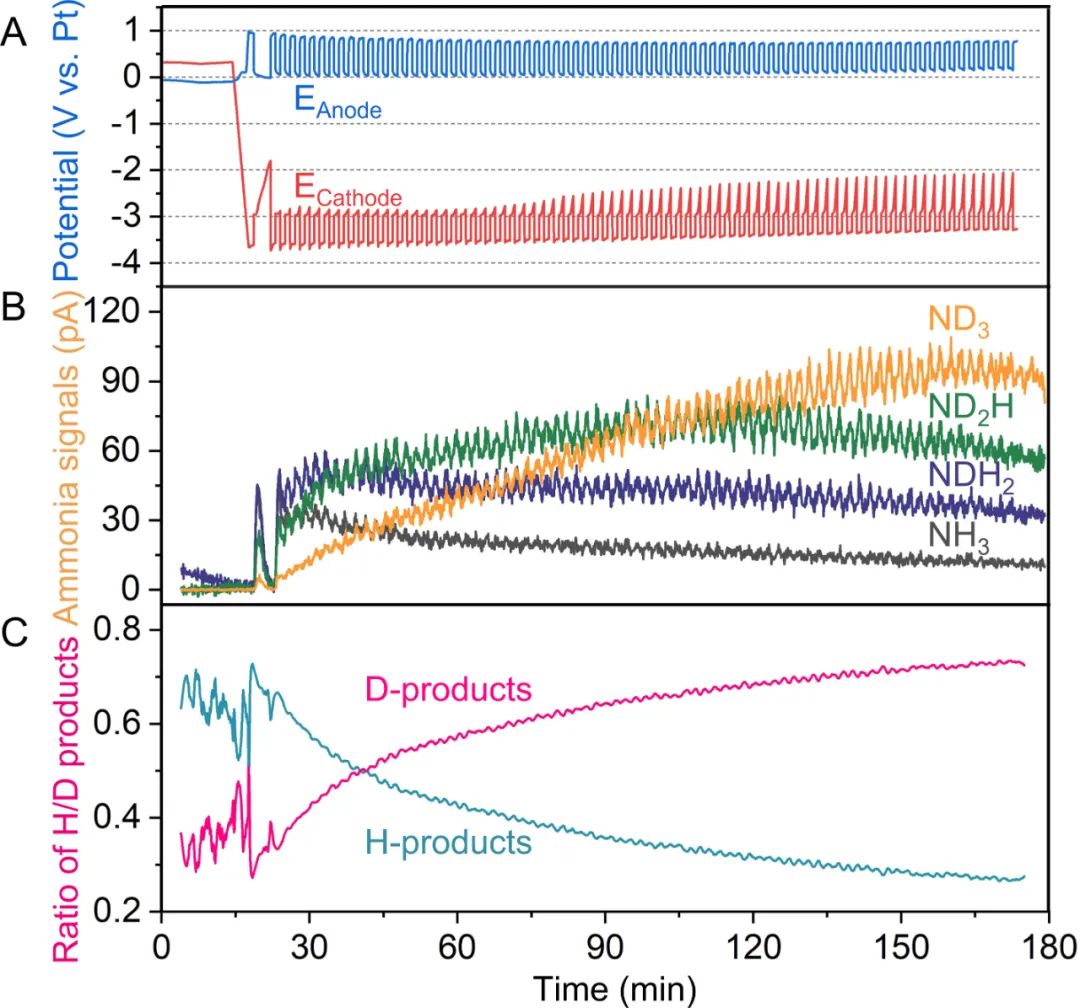
图3. 同位素标记(D2氧化)原位质谱分析
采用电位循环(potential cycling)策略提高Li-NRR的性能,即在施加电位和开路电压(OCV)之间切换电位。改变电位循环时间的实验表明,最佳电位循环的条件是 1 min进行锂沉积,1 min进行开路电压(1 min deposition + 1 min resting)。如图4 A所示,阴极的静息电位大约是-2 V vs. Pt。
采用模数电池威廉希尔官方网站
(modulo battery technique)可以控制静息电位,改变静息电位的实验表明,最佳的静息电位是-2 V vs. Pt。Li-NRR与阳极的HOR耦合,使得平均阳极电位保持在0.6 V 左右,极大降低了电解池的槽电压(4.3 V)。在连续流电解池中评估质子穿梭剂(EtOH)浓度对性能的影响(图4 B),最佳EtOH浓度为0.25 vol.%。
在最优条件下,使用30 μm SSC实现了61 ± 1%的产氨FE。电位循环的促进作用在质子和氮气扩散限制区表现出相反的作用。这符合理论模型分析,在质子限制区,在电位循环的静息期,沉积的锂金属与N2反应生成氮化锂,氮化锂溶解之后释放NH3,从而提高性能。
在N2限制区,沉积的锂金属与质子反应形成氢化锂,氢化锂溶解产生H2,导致产氨的选择性降低。所产的氨主要分布在气相、电解液和电极沉积物中,最佳条件下,大约50%的氨分布在气相中,这有利于后期分离和富集氨(图4 C)。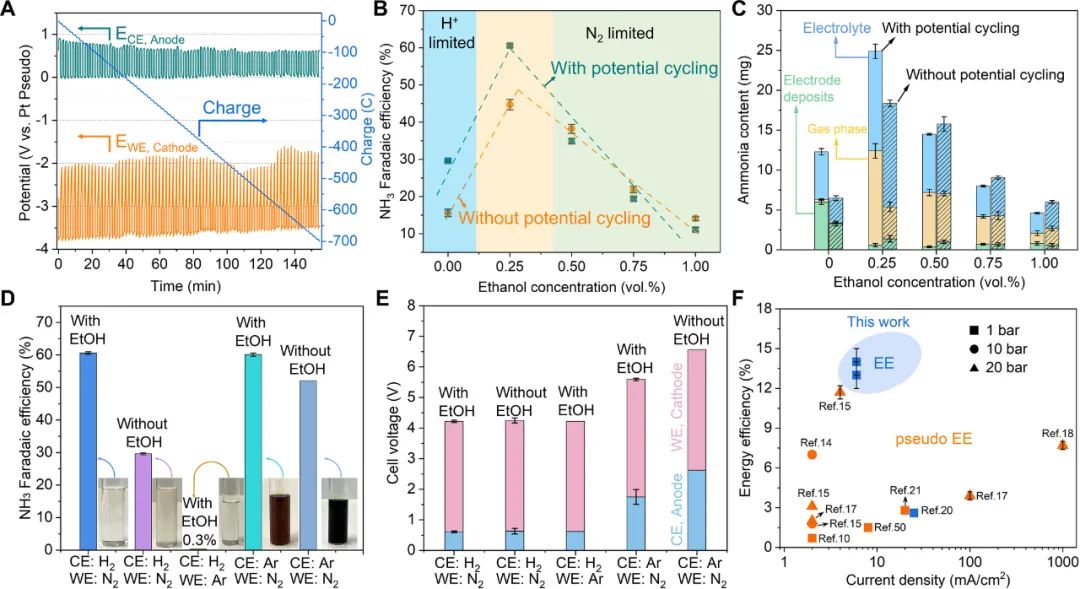
图4. 电位循环法和质子/氮气限制区域的权衡策略提高锂介导合成氨的性能
对照实验进一步确认HOR对阳极电位降低的重要性(图4 D和E)。在典型的实验条件下(阳极通入H2,阴极通入N2,使用EtOH作为质子穿梭剂),测试后的电解液无色透明,阳极电位为0.6 V。其他条件相同时,当在阳极测通入Ar时,测试之后的深色电解液表明有机溶剂严重的氧化和分解,阳极电位高达1.7 V (有EtOH)和2.6 V(无EtOH),这清晰地表明HOR可保护电解液不被氧化,降低了阳极电位。
采用NMR分析反应前后电解液的变化也支持了这一结论。图4 F总结了电流密度和能量效率(EE),需要指出的是,伪能量效率未使用氢气或者所计算的EE未包含产氢所需的能量。这项工作着重证明了在阳极引入HOR可大大降低阳极电位,证明了所产氨中的氢来自于HOR,不是牺牲有机溶剂。
还强调了整个系统能量效率以及在阳极上引入HOR提供可持续氢源的重要性。未来的工作应侧重于在更高的电流密度下获得高EE和FE,以提高产的氨速率。 只有当阳极真正使用氢气或水作为可持续的氢源时,实现高FE和电流密度才具有实际的意义。
尽管该研究开发了有机体系中HOR高稳定性的PtAu催化剂,提高了流动电解池的运行稳定性,实现了连续化电化学合成氨,但是这项工作并未解决所有在工业应用层面的锂介导合成氨的问题。未来的Li-NRR研究应该着眼于提高电流,优化H2/N2的传质以及精确调节GDE上气体和液体之间的压力梯度。
主要目标应该是在中试规模的连续流电解池中,在工业电流密度条件下,实现高的FE和EE。可持续的氢气可以通过可再生电力驱动的电解水获得。循环阳极出口H2可能是提高阳极H2利用效率的途径之一。本研究的这些发现为锂介导合成氨的进一步发展提供了坚实的基础和指导。
审核编辑:刘清
-
电解电容
+关注
关注
14文章
672浏览量
50851 -
ssc
+关注
关注
0文章
24浏览量
11207 -
电解池
+关注
关注
0文章
24浏览量
9517
原文标题:Science:氮气还原耦合氢气氧化实现连续流电化学合成氨
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
电化学气体传感器信号放大调试经验
扫描速率对各体系的电化学行为有什么影响
哈尔滨工业大学/南方科技大学:聚焦离子束制备高分辨率电化学-电致发光耦合双极纳米电极阵列传感器

电化学测试方法详解
电化学储能和电池储能的关系
电化学储能与物理储能的对比
电化学储能系统的组成与作用
电化学储能的基本原理介绍
深度解析电化学储能最新官方数据






 工商网监
工商网监
评论